新薬レビュー2022
米国の製薬会社。 2022;47(10):24-30.
FDA の定義によると、新分子エンティティ (NME) は、米国で初めて上市された化学物質を有効成分として含む新しい医薬品です。 2021 ~ 2022 年に承認された NME の次の説明 ( 表1 ) は、各新薬の基本的な臨床的および薬理学的プロファイル、ならびに重要な注意事項と警告を詳述しています。また、メーカーの新薬申請をサポートするために FDA に提出された選択された薬物動態、副作用、薬物相互作用、および投薬データの簡単な要約も含まれています。このレビューは、内容を評価するものではなく、客観的なものにすることを目的としています。各 NME の情報は、主に FDA の承認前に公開された情報源から入手したものです。経験は、NME の治療プロファイルの多くの側面が市販前の臨床研究では検出されず、より広範な集団で薬物が使用された後に明らかになることを明確に示しています。たとえば、一部の NME では、これまで報告されていなかった副作用が、市販後数年以内に明らかになります。一部の NME は最終的に、重篤な薬物有害反応について少なくとも 1 つのブラック ボックス警告を取得するか、承認時に認識されなかった安全上の理由により市場から撤退する可能性があります。したがって、このレビューはいくつかの新薬の紹介を提供しますが、開業医は、製薬文献や患者によって報告されている治療プロファイルの変化を認識することが不可欠です.
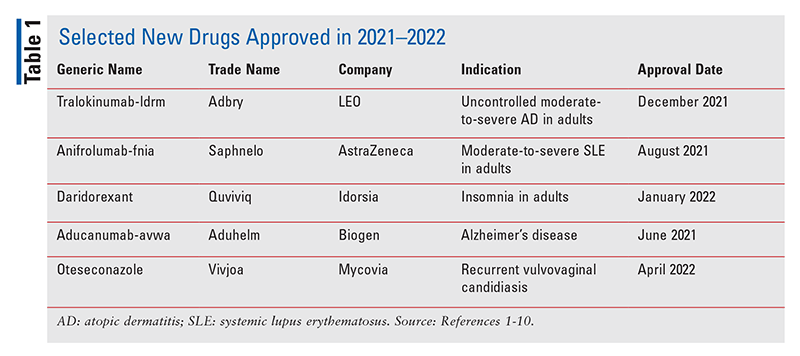
Tralokinumab-ldrm (アドブリー、LEO)
適応と臨床プロファイル 1.2 : FDA は、中等度から重度のアトピー性皮膚炎 (AD) の治療薬としてトラロキヌマブ-ldrm を承認しました。成人のアトピー性皮膚炎 (AD) は、外用薬で十分に制御されていないか、またはそれらの治療法が推奨されていません。 AD、別名 アトピー性湿疹 は、皮膚の乾燥、かゆみ(特に夜間)、および体のさまざまな部分の赤から茶色がかった灰色の斑点を特徴とする、慢性的に再発する炎症性皮膚疾患です。 AD の発生率は先進国ではほぼ 3 倍に増加しており、世界中の子供の約 15% から 20%、成人の 1% から 3% に影響を与えています。トラロキヌマブ-ldrm は、AD の管理のために承認された最初の生物学的薬剤であり、この疾患の重要な炎症メディエーターであるインターロイキン (IL)-13 を阻害することによって機能します。 Tralokinumab-ldrm は、局所コルチコステロイドの有無にかかわらず使用できます。
この薬剤の承認は、1,500 人以上の成人患者を対象とした 3 つの二重盲検プラセボ対照多国籍第 III 相試験 (ECZTRA 1、ECZTRA 2、および ECZTRA 3) の有効性と安全性の結果に基づいています。患者は、トラロキヌマブ-ldrm 300 mg を隔週で単独または局所コルチコステロイドと一緒に注射されました。主要評価項目は、16 週で 0 または 1 (IGA 0/1) の治験責任医師の総合評価スコア、および 16 週で湿疹領域および重症度指数 (EASI 75) の 75% 以上の改善でした。IGA 0/1 および/または16 週目にトラロキヌマブ-ldrm を投与した EASI 75 は、2 週間または 4 週間ごとにトラロキヌマブ-ldrm を投与するか、36 週間までプラセボを投与するように再無作為化されました。 16 週目に、プラセボ患者よりも多くのトラロキヌマブ ldrm 患者が IGA 0/1 (38.9% 対 26.2%) および EASI 75 (56.0% 対 35.7%) を達成しました。 16 週目の応答者のうち、2 週間ごとにトラロキヌマブ ldrm で治療された患者の 89.6% と 92.5%、4 週間ごとにトラロキヌマブ ldrm で治療された患者の 77.6% と 90.8% が、それぞれ IGA 0/1 と EASI を維持しました。 32 週で 75。16 週でトラロキヌマブ-ldrm で IGA 0/1 と EASI 75 を達成しなかった患者では、それぞれ 30.5% と 55.8% が 32 週でこれらのエンドポイントを達成しました。
薬理学と薬物動態 1.2 : Tralokinumab-ldrm は、ヒト免疫グロブリン (Ig) G4 モノクローナル抗体であり、ヒト IL-13 に特異的に結合し、この受容体の α-1 および α-2 サブユニットとの相互作用を阻害します。典型的には 2 型免疫応答によって産生されるサイトカインである IL-13 は、AD に関与する炎症誘発性サイトカイン、ケモカイン、および IgE の放出を促進します。
トラロキヌマブ-ldrm の絶対バイオアベイラビリティは 76% と推定され、血漿濃度が最大になるまでの時間 (T 最大 ) 投与後 5 ~ 8 日。分布量は 4.2 L と推定されます。Tralokinumab-ldrm はペプチド代謝を受けるようです。半減期は 3 週間で、全身クリアランスは 0.149 L/日と推定されます。
有害反応と薬物相互作用 1.2 : 臨床試験中にトラロキヌマブ-ldrm で報告された一般的な有害事象には、好酸球増多症、眼とまぶたの炎症、上気道感染症、および注射部位反応が含まれます。患者は、結膜炎や角膜炎などの目の症状が新たに発症したり悪化したりした場合は、医療提供者に報告することをお勧めします。アナフィラキシーを含む深刻な過敏反応と血管性浮腫が発生しており、投薬の中止が必要です。 Tralokinumab-ldrm は、駆虫療法の有効性を損なう可能性があります。したがって、既存の蠕虫感染症のある患者や治療中に感染した患者では、開始または継続すべきではありません。トラロキヌマブと ldrm による治療中は、生ワクチンの使用を避ける必要があります。
薬物相互作用に関しては、ミダゾラム (CYP3A4 基質)、ワルファリン (CYP2C9 基質)、オメプラゾール (CYP2C19 基質)、メトプロロール (CYP2D6 基質)、およびカフェイン (CYP1A2 基質) の薬物動態に対するトラロキヌマブ-ldrm の効果を評価する小規模な臨床試験が観察されました。曝露に有意な変化なし (AUC または C 最大 )トラロキヌマブを複数回投与した後の CYP450 基質の変化を、トラロキヌマブ-ldrm の単独投与と比較しました。
投薬と管理 1.2 : Tralokinumab-ldrm は、SC 注射用に 150 mg/mL プレフィルド シリンジで入手できます。推奨用量は、最初の用量として 600 mg (150 mg の注射を 4 回)、その後 300 mg (150 mg の注射を 2 回) を隔週で投与します。 16 週間の治療後、体重が 100 kg 未満で透明またはほぼ透明の皮膚を達成している患者では、4 週間ごとに 300 mg の投与量を考慮することができます。現在の予防接種ガイドラインで推奨されているように、すべての年齢に応じた予防接種は、治療開始前に完了する必要があります。
Anifrolumab-fnia (サフネロ、アストラゼネカ)
適応と臨床プロファイル 3.4 : Anifrolumab-fnia は、標準治療を受けている成人の中等度から重度の全身性エリテマトーデス (SLE) の治療薬として承認されている I 型インターフェロン (IFN) 受容体拮抗薬です。 anifrolumab-fnia の安全性と有効性は、MUSE、TULIP-1、および TULIP-2 の 3 つの多国籍プラセボ対照臨床試験で確立されました。 MUSE は第 II 相試験であり、主要評価項目には SLE Responder Index (SRI-4) の評価と 24 週目に測定された経口コルチコステロイド (OCS) の持続的減少が含まれていました。 -fnia 300 mg、アニフロルマブ-fnia 1,000 mg、またはプラセボ。第 III 相試験である TULIP-1 および TULIP-2 試験では、52 週で評価された疾患活動性の改善という主要な結果が得られました。チューリップ-2。 TULIP-1 および TULIP-2 の副次的アウトカムには、OCS 減少の維持、皮膚 SLE 活動の改善、およびフレア率が含まれていました。
52 週目の BICLA 奏効率は 3 つの試験すべてで報告されましたが、主要評価項目は TULIP-2 のみでした。回答率の差は、MUSE で 28.8%、TULIP-1 で 17%、TULIP-2 で 16.3% でした。 52 週目の SRI-4 反応率は、MUSE で 24%、TULIP-1 で 6%、TULIP-2 で 18.2% でした。 Anifrolumab-fnia は、TULIP-1 の応答率に統計的に有意な差を示しませんでした。
薬理学と薬物動態 3.4 : Anifrolumab-fnia は、I 型 IFN 受容体 (IFNAR1) のサブユニット 1 に結合し、その活性を阻害する免疫グロブリン (Ig) G1-κ モノクローナル抗体です。 I型IFNの活性が低下すると、SLEの病因におけるその役割が低下します。 Anifrolumab-fnia は、IFNAR1 を内在化するようにも作用し、利用可能な受容体を減少させます。これらの作用機序は、SLEの病理に寄与する炎症および免疫学的プロセスの減少をもたらします。
Anifrolumab-fnia は、4 週間ごとに 100 mg から 1,000 mg の用量範囲で非線形の薬物動態を示し、用量が増加するにつれて AUC 曝露が増加します。典型的な SLE 患者 (69.1 kg) の分布量は 6.23 L と推定されます。標準的な投与量に基づいて、アニフロルマブ-fnia 300 mg IV の 4 週間ごとのクリアランスは 0.193 L/日と推定されました。
有害反応と薬物相互作用 3.4 : anifrolumab-fnia による最も一般的な (>10%) 有害反応は、インフルエンザ、尿路感染症、結核、気管支炎、および (34% の発生率) 上気道感染症 (すなわち、鼻咽頭炎、咽頭炎) を含む感染症 (患者の 70%) でした。 、および副鼻腔炎)。治療中の重篤な感染症の発生率は 5% でした。現在、ヒトの妊娠におけるアニフロルマブ-fniaの使用に関するデータは限られており、主要な先天性欠損症、流産、または母体または胎児の有害転帰の薬物関連リスクについて情報を提供するには不十分です。モノクローナル IgG 抗体は、妊娠が進むにつれて胎盤を介して活発に輸送されることが知られています。したがって、アニフロルマブ-fniaへの胎児の曝露は、妊娠第3期に大きくなる可能性があります。
唯一報告されている anifrolumab-fnia の禁忌は、anifrolumab またはその製剤の成分に対する重度の過敏症 (アナフィラキシーなど) です。懸念される臨床的に顕著な副作用は、重篤な感染症、アナフィラキシーを含む過敏症反応、および悪性腫瘍です。今日まで、正式な薬物相互作用研究は実施されていません。
投薬と管理 3.4 : Anifrolumab-fnia は、300 mg/2 mL (150 mg/mL) を含む単回投与バイアルとして提供されます。推奨投与量は、30 分かけて 300 mg を IV 注入として投与することです。投与前に、無菌技術を使用してアニフロルマブ-fnia を 0.9% 塩化ナトリウム注射液 (USP) で希釈する必要があります。 2 mL の 0.9% 塩化ナトリウム注射液を 100 mL の輸液バッグから取り出し、2 mL の anifrolumab-fnia を穏やかに混合しながら加えて、総量を 100 mL にします。輸液は、無菌の低タンパク質結合 0.2 または 0.22 ミクロンのインライン フィルターを含む輸液ラインを介して、調製直後に 30 分間かけて投与する必要があります。注入が完了したら、25 mL の 0.9% 塩化ナトリウム注射液 (USP) でラインをフラッシュする必要があります。
Daridorexant (Quviviq、Idorsia)
適応と臨床プロファイル 5.6 : ダリドレキサントは、入眠困難および/または睡眠維持困難を特徴とする成人不眠症の治療薬として承認されました。最も一般的な睡眠障害である不眠症 (米国の成人の 30% 以上に発生) は、十分な睡眠を得ることが困難であり、睡眠に対する不満と日中の機能への重大な悪影響が組み合わさったものと定義されています。慢性不眠症は、少なくとも 3 か月間、週に 3 日以上の睡眠を開始および/または維持することが困難であると見なされます。不眠症は、覚醒に関連する脳の領域の過剰活動に起因すると考えられ、患者の気分、エネルギーレベル、および集中力に重大な影響を及ぼします。さらに、長期にわたる不眠症は、精神障害、心血管疾患、2 型糖尿病、薬物乱用、認知症などの深刻な健康状態と関連しています。
daridorexant の有効性は、研究 1 と研究 2 の 2 つの同等に無作為化された多施設プラセボ対照試験で評価されました。 精神障害の診断と統計マニュアル 、第 5 版では、3 か月間、1 日 1 回夕方に daridorexant またはプラセボを投与されました。研究 1 では、930 人の被験者が daridorexant 25 mg、daridorexant 50 mg、またはプラセボに割り当てられました。研究 2 では、924 人の被験者が daridorexant 25 mg、daridorexant 10 mg、またはプラセボに割り当てられました。 3 か月の治療期間の終わりに、両方の研究に 7 日間のプラセボの実行期間が含まれ、その後、被験者は延長研究に入ることができました。合計 600 人の被験者が少なくとも 6 か月間治療を受けました。これらのうち、373 人が少なくとも 12 か月間治療を受けました。両方の研究の主要な有効性エンドポイントは、ベースラインから 1 か月および 3 か月までの持続的な睡眠 (LPS、入眠時間) および入眠後の覚醒 (WASO、睡眠維持) の変化であり、睡眠研究所での睡眠ポリグラフによって客観的に測定されました。研究 1 では、ダリドレキサント 25 mg とダリドレキサント 50 mg は、1 か月目と 3 か月目の LPS と WASO、および自己申告の総睡眠時間 (sTST) において、プラセボと比較して統計的に有意な改善を示しました。主要な副次評価項目である 1 か月目と 3 か月目の不眠症の日中の症状と影響アンケート 7 の眠気ドメイン スコアで測定した眠気とプラセボの比較。研究 2 では、1 か月目と 3 か月目の時点で 25 mg の用量がプラセボに対して統計的に有意な改善を示しましたが、10 mg の用量ではそうではありませんでした。
薬理学と薬物動態 5.6 : オレキシン A とオレキシン B は、主に外側視床下部のニューロンから分泌される神経ペプチドです。これらの神経ペプチドは、オレキシン受容体タイプ 1 とオレキシン受容体タイプ 2 (OX1R、OX2R) の 2 つの G タンパク質共役受容体 (GPCR) に結合して活性化することにより、その効果を引き出します。オレキシン受容体経路は、睡眠覚醒リズムを含む多くの生理学的プロセスにおいて重要な調節的役割を果たしています。ダリドレキサント ( 図1 ) は、OX1R と OX2R のデュアル アンタゴニストとして機能するように設計されたベンズイミダゾール-ピロリジンであり、それによってオレキシン A とオレキシン B の覚醒促進活性を抑制します。

daridorexant の経口バイオアベイラビリティは 62% で、血漿濃度のピークは 1 ~ 2 時間以内に発生します (T 最大 )。ダリドレキサントの分布容積は 31 L で、99.7% が血漿タンパク質に結合しています。 Daridorexant は、主に CYP3A4 (89%) によって広範な代謝を受けます。排泄の主な経路は糞便 (57%) です。主に代謝物として尿中に排泄される割合はわずかです (28%)。 daridorexant の最終半減期は約 8 時間です。ダリドレキサントは入眠時に急速に吸収され、夜の睡眠後に 80% が排出されるクリアランス プロファイルにより、残留効果を最小限に抑えることができます。
有害反応と薬物相互作用 5.6 : 臨床試験で daridorexant に関連する最も一般的な副作用 (>5%、およびプラセボよりも多い) は、頭痛、傾眠または疲労でした。製品ラベルには、うつ病や自殺念慮の悪化、夢遊病、睡眠運転などの複雑な睡眠行動、完全に起きていないときの他の活動 (例: 食事の準備/食事、電話をかける、セックスなど) に関する警告も含まれています。 .これらの副作用のいずれかが発生した場合、患者はすぐに医療提供者に連絡する必要があります。患者が入眠時または覚醒時に、最大数分間持続する幻覚および睡眠麻痺 (一時的に動くことも話すこともできない) が発生することがあります。ダリドレキサントは、ナルコレプシー患者には禁忌です。
重大な先天性欠損症、流産、またはその他の母体または胎児への有害転帰の薬物関連リスクを決定するための、妊娠中の女性におけるダリドレキサントの使用に関するデータはありません。動物の生殖研究では、ダリドレキサントは、ヒトの最大推奨用量の 8 ~ 10 倍までの用量で胎児毒性を引き起こしませんでした。母乳中のダリドレキサントの存在、母乳育児への影響、または乳生産への影響に関するデータは存在しませんが、授乳中のげっ歯類の乳から薬物とその代謝物が検出されました。したがって、母乳を介してダリドレキサントにさらされる可能性のある乳児は、過度の鎮静を監視する必要があります。ダリドレキサントは、乱用される可能性があり、依存につながる可能性があるため、連邦政府が規制する物質です。
投薬と管理 5.6 : Daridorexant は、経口投与用の 25 mg および 50 mg のフィルムコーティング錠として提供されます。推奨用量は、就寝前 30 分以内に 25 ~ 50 mg を 1 晩 1 回、起床予定時刻まで少なくとも 7 時間空けて服用することです。 daridorexant を食事と一緒に、または食事の直後に服用すると、入眠までの時間が遅くなる可能性があります。 Daridorexant は、他の中枢神経抑制薬やアルコールと一緒に服用しないでください。また、薬物の影響を受けている間は、運転、重機の操作、または明確な思考を必要とする活動を行うことを避けるように患者にアドバイスする必要があります。中等度の肝障害のある患者では、最大推奨用量は 25 mg で、1 晩に 1 回までです。ダリドレキサントは、重度の肝障害のある患者には推奨されません。 daridorexant と CYP3A4 の強力な阻害剤との併用は避けるべきであり、中等度の CYP3A4 阻害剤との併用療法を受けている患者では、用量を 25 mg に制限する必要があります。 daridorexant と中程度から強力な CYP3A4 インデューサーの併用も避けるべきです。
Aducanumab-avwa (Aduhelm、バイオジェン)
適応と臨床プロファイル 7.8 : Aducanumab-avwa は、アルツハイマー病の治療が適応となるアミロイド ベータ (AB) に対する抗体であり、2003 年以来、この疾患に対する最初の新しい治療法です。重篤または生命を脅かす疾患に対する既存の治療法よりも優れた治療上の利点。この適応症の継続的な承認は、確認試験で臨床的利益が検証されることを条件としています。アルツハイマー病は、620 万人のアメリカ人が罹患している不可逆的で衰弱性があり、進行性の脳障害であり、記憶、思考、そして最終的には単純なタスクを実行する能力さえもゆっくりと侵食します。具体的な原因は完全にはわかっていませんが、アルツハイマー病は、アミロイド斑や神経原線維変化 (タウ) などの脳の変化を特徴とし、記憶や思考を調節する重要な脳領域のニューロンが失われます。現在利用可能な治療法は疾患の症状のみを治療しますが、aducanumab-avwa は、アルツハイマー病の根底にあると推定されるプロセスを標的にして変更する最初の薬剤です。
aducanumab-avwa の早期承認は、アルツハイマー病の軽度認知障害または軽度認知症段階の合計 3,482 人の患者を含む 3 つの二重盲検プラセボ対照用量範囲試験の結果に基づいています。 EMERGE (Study 1)、ENGAGE (Study 2)、および PRIME (Study 3) として知られる研究では、アルツハイマー病の病態に関与している脳領域と、アルツハイマー病に関与していない脳領域の AB プラークを定量化するために陽電子放出断層撮影法が使用されました。 aducanumab-avwaを投与された患者では、ABプラークが用量依存的および時間依存的に大幅に減少しました(研究全体で59%〜71%)が、対照ではABプラークは減少しませんでした。今日まで、アルツハイマー病の早期または後期段階での aducanumab-avwa 治療の開始に関する安全性または有効性のデータはありません。
薬理学と薬物動態 7.8 : Aducanumab-avwa は、血液脳関門を通過して、脳内の AB プラークの凝集した可溶性オリゴマーと不溶性原線維コンフォメーションを選択的に標的にして結合できるヒト免疫グロブリン-ガンマ (IgG) 1 モノクローナル抗体です。生化学的研究は、aducanumab-avwa が AB アミノ酸 3 ~ 7 によって形成される線形エピトープに選択的に結合し、それによって他のタンパク質との相互作用を最小限に抑えることを示しています。 aducanumab-avwa は、弱い一価親和性、速い結合速度、およびエピトープに富む凝集体に対する強い結合力に基づいて、AB 単量体とオリゴマーまたは線維状凝集体を区別することが示されています。
aducanumab-avwa の定常状態濃度は、4 週間ごとのレジメンによる 16 週間の反復投与によって達成されます。ピーク濃度 (C 最大 )、トラフ濃度 (C 分 )、および定常状態での血漿濃度対時間曲線下面積は、標準投与範囲(4週間ごとに1〜10 mg / kg)にわたって用量比例的に増加します。定常状態での推定分布量は 9.6 L です。内因性 IgG と同様に、aducanumab-avwa は、タンパク質異化経路によって小さなペプチドに加水分解され、最終的にはアミノ酸になると推定されます。 aducanumab-avwa の最終半減期は約 25 日です。腎障害または肝障害のある患者の薬物動態を評価した研究は報告されていませんが、この薬物は腎排泄または肝代謝を受けるとは予想されません。体重、年齢、性別、および人種の違いにより、薬物曝露が変化する可能性がありますが、臨床反応に大きな影響を与えることはわかりませんでした.
有害反応と薬物相互作用 7.8 : aducanumab-avwa で最もよく報告されている副作用には、アミロイド関連の画像異常(ARIA)が含まれます。頭痛;落ちる;下痢;混乱、見当識障害、および精神状態の変化。製品ラベルには、脳の一部の一時的な腫れとして最も一般的に現れる ARIA の警告が含まれています。通常、この影響は時間の経過とともに解消され、症状を引き起こすことは知られていません。しかし、一部の ARIA 患者は、aducanumab-avwa に起因する頭痛、錯乱、めまい、視力の変化、または吐き気を報告しました。まれに、aducanumab-avwa の使用が、血管性浮腫や蕁麻疹などの過敏反応と関連しています。
投薬と管理 7.8 : Aducanumab-avwa は、170 mg/1.7 mL (100 mg/mL) および 300 mg/3 mL (100 mg/mL) の単回投与バイアルで、防腐剤を含まない IV 注射用の無菌溶液として提供されます。最初の滴定 (6 週間以上) 後、推奨用量は 10 mg/kg を 4 週間ごとに約 1 時間、少なくとも 21 日間隔で IV 注入します。最初の滴定には、注入 1 および 2 の 1 mg/kg 用量の投与が含まれます。注入 3 および 4 では 3 mg/kg の用量。輸液 5 および 6 では 6 mg/kg の用量。腎臓または肝臓の障害に対する用量調整は想定されていません。
オテセコナゾール(Vivjoa、Mycovia)
適応と臨床プロファイル 9.10 : オテセコナゾールは、生殖能力のない RVVC の病歴を持つ女性における再発性外陰膣カンジダ症 (RVVC) の発生率を減らすために特別に指示された最初で唯一の薬剤です。その使用は、妊娠中および授乳中の女性には禁忌です。一般に慢性イースト菌感染症として知られる RVVC は、12 か月間に 3 回以上の急性イースト菌感染症の症状として定義されます。症状には、膣のかゆみ、灼熱感、刺激、炎症が含まれ、一部の患者は、異常なおりものや、排尿時または性交時の痛みを経験します。成人女性の 75% が生涯に少なくとも 1 回イースト菌に感染し、約 50% が再発すると推定されています。これらの女性のうち、最大 90% が RVVC を発症します。 FDA はオテセコナゾールをファストトラックおよび認定感染症製品に指定しました。
オテセコナゾールは、11 カ国の 232 施設からの 875 人の患者を含む 3 つの第 III 相臨床試験の有効性データに基づいて承認されました。 2 つの国際的な VIOLET 研究では、オテセコナゾールを投与された RVVC の女性の 93% から 96% が 48 週間の維持期間中に再発しませんでした。対照的に、プラセボ投与者の 57% から 61% は再発を経験しませんでした。米国を拠点とするウルトラバイオレット研究では、オテセコナゾールを投与された RVVC の女性の 89.7% が最初のイースト菌感染症を解消し、50 週間の維持期間中に再発していませんでしたが、フルコナゾールとプラセボを投与された女性の 57.1% は再発しませんでした。
薬理学と薬物動態 9.10 : オテセコナゾール ( 図 2 ) は、14-α-デメチラーゼ (CYP51) を阻害する新規テトラゾール抗真菌誘導体であり、必須真菌ステロール エルゴステロールの生合成の初期段階を触媒する真菌酵素です。さらに、14-α-デメチラーゼの阻害は、真菌にとって有毒な 14-メチル化ステロールの蓄積をもたらします。テトラゾール金属結合基がその構造に組み込まれているため、オテセコナゾールはヒト CYP 酵素に対する親和性が低下し、おそらく、有害反応および薬物相互作用プロファイルが改善されています。

オテセコナゾールの血漿濃度がピークに達するまでの時間は、5~10時間の範囲です。高脂肪、高カロリーの食事での投与はCを増加させました 最大 とAUC 0-72h それぞれ 45% と 36% 増加しましたが、低脂肪、低カロリーの食事では有意差は見られませんでした。オテセコナゾールの分布量は約 423 L で、99% 以上が血漿タンパク質に結合しています。動物研究は、膣組織におけるオテセコナゾール曝露が血漿曝露に匹敵することを示しています。オテセコナゾールは有意な代謝を受けません。経口投与量の約 56% が胆汁排泄によって糞便中に排泄され、26% が尿中に排泄されます。オテセコナゾールの半減期の中央値は約 138 日です。
有害反応と薬物相互作用 9.10 : 最も頻繁に報告された副作用は頭痛と吐き気で、試験参加者のそれぞれ 7.4% と 3.6% で発生しました。臨床試験で 2% 未満の患者で報告された有害反応には、消化不良、のぼせ、排尿障害、生殖器系障害 (月経過多、子宮出血、外陰腟刺激)、および血中クレアチンホスホキナーゼの増加が含まれます。オテセコナゾールは、動物実験で胎児に害を及ぼすことが判明しました。したがって、妊娠中および授乳中の女性だけでなく、生殖能力のある女性には禁忌です。オテセコナゾールは、この薬に対する既知の過敏症のある患者にも禁忌です。
オテセコナゾールは乳癌耐性タンパク質 (BCRP) 阻害剤であり、ロスバスタチンなどの BCRP 基質と併用すると、基質の露出が増加し、これらの薬剤に関連する有害反応のリスクが高まる可能性があります。したがって、BCRP基質がオテセコナゾールと同時に投与される場合、BCRP基質の最小有効初期用量および維持用量を使用することが推奨され、患者は副作用について監視されるべきである.
投薬と管理 9.10 : オテセコナゾールは、経口投与用の 150 mg ハード ゼラチン カプセルとして入手できます。 1) オテセコナゾール単独、および 2) オテセコナゾールとフルコナゾールの 2 つの推奨される投与計画があります。製品ラベルには、これらのレジメンの詳細な投与スケジュールが記載されています。軽度から中等度の腎障害または軽度の肝障害のある患者では、用量調整は推奨されません。ただし、オテセコナゾールは、重度の腎障害または末期腎疾患(透析の有無にかかわらず)の患者、または中等度または重度の肝障害のある患者には推奨されません。
参考文献
1. Adbry (tralokinumab-ldrm) 添付文書。ニュージャージー州マディソン:LEO Pharma Inc。 2022 年 7 月。
2. Wollenberg A、Howell MD、Guttman-Yassky E、他。抗IL-13 mAbであるトラロキヌマブによるアトピー性皮膚炎の治療。 Jアレルギークリニック免疫 . 2019;143(1):135-141.
3. サフネロ(anifrolumab-fnia)の添付文書。デラウェア州ウィルミントン:AstraZeneca Pharmaceuticals LP。 2022 年 7 月。
4. アニフロルマブ:医薬品情報。最新の。マサチューセッツ州ウォルサム: UpToDate; 2022. www.uptodate.com. Accessed June 28, 2022.
5. Quviviq (daridorexant) 添付文書。ペンシルバニア州ラドナー: Idorsia Pharmaceuticals US Inc; 2022 年 4 月。
6. Markham A. Daridorexant: 最初の承認。 薬物 . 2022;82(5):601-607.
7. Aduhelm (aducanumab-avwa) 添付文書。マサチューセッツ州ケンブリッジ:バイオジェン社。 2021 年 6 月。
8. Sevigny J、Chiao P、Williams Lなど。前駆期または軽度のアルツハイマー病患者における抗アミロイドベータモノクローナル抗体である Aducanumab (BIIB037): 無作為化、二重盲検、プラセボ対照、第 1b 相試験の中間結果。 アルツ認知症 . 2015;11(7 補足 6):P277.
9. Vivjoa(オテセコナゾール)の添付文書。ノースカロライナ州ダーラム: Mycovia Pharmaceuticals, Inc; 2022 年 4 月。
10. Sobel JD、Nyirjesy P. Oteseconazole: 再発性外陰膣カンジダ症の治療の進歩。 未来の微生物 . 2021;16(18):1453-1461.
この記事に含まれる内容は、情報提供のみを目的としています。このコンテンツは、専門家のアドバイスに代わるものではありません。この記事に記載されている情報を信頼することは、ご自身の責任で行ってください。











